Cart
×
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Oct. 4, 2017– Catabasis Pharmaceuticals, Inc. (NASDAQ:CATB), a clinical-stage biopharmaceutical company, today reported new positive efficacy results showing sustained disease-modifying effects in the MoveDMD trial open-label extension following 24 and 36 weeks of treatment with edasalonexent. Across all key assessments of muscle function, improvements were observed in the rate of decline after 24 and 36 weeks of oral 100 mg/kg/day edasalonexent treatment compared to the rate of change in the control period for boys prior to receiving edasalonexent treatment. These data provide clinically meaningful evidence that edasalonexent substantially slowed the progression of Duchenne muscular dystrophy (DMD).
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20171004005218/en/
Additionally, supportive changes in measures of muscle health were seen, consistent with positive edasalonexent treatment effects. Muscle enzymes significantly decreased compared to baseline at 12 weeks and later time points (p<0.05) and lower leg muscle MRI T2 rate of change was significantly improved in comparison to progression during the control period (p≤0.05). Edasalonexent continued to be well tolerated with no safety signals observed in the trial.
Based on the consistency of the MoveDMD results and supportive regulatory input from FDA, Catabasis plans to initiate a single global Phase 3 trial with edasalonexent in patients with DMD regardless of mutation type in the first half of 2018 with top-line results expected in 2020. The data were presented today in a late breaking session at the World Muscle Society Conference and will be discussed, along with the Phase 3 clinical trial plan, on a conference call today, October 4, 2017.
“We are extremely excited to see edasalonexent change the trajectory of disease in the MoveDMD trial with substantially slowed disease progression,” said Jill C. Milne, Ph.D., Chief Executive Officer of Catabasis. “Boys treated with edasalonexent stabilized; they experienced meaningful improvements in muscle function compared to the rates of change observed during the control period. Importantly, other supportive positive measures of muscle health were observed. We look forward to advancing edasalonexent as a disease-modifying therapy in a single Phase 3 pivotal trial as soon as possible with the goal of providing a meaningful impact on disease progression for all boys affected by Duchenne.”
“Our goal in treating boys with Duchenne is to slow the progression of the disease. It is tremendously encouraging to see boys taking edasalonexent stabilize in their functional abilities and MRI T2 measures along with its continued safety profile,” said Richard Finkel, M.D., Chief, Division of Neurology, Department of Pediatrics at Nemours Children’s Health System and a Principal Investigator for the study. “I look forward to continuing to investigate edasalonexent as a potential therapy for the many boys affected by this devastating disease.”
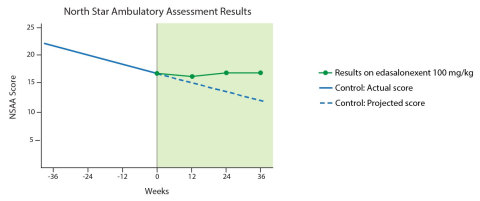
In the MoveDMD trial, a substantial slowing of the disease progression of DMD was seen in boys treated with edasalonexent compared to the rates of change during the control period. Through 36 weeks of treatment, the 100 mg/kg/day treatment group showed clinically meaningful numerical improvements in rates of decline compared to rates of change during the control period across all three timed function tests (10-meter walk/run, 4-stair climb and time to stand), as well as the North Star Ambulatory Assessment (NSAA), an integrated global assessment of muscle function. Control period changes were measured prior to boys receiving edasalonexent, either prior to Phase 2 or in the placebo group, for time periods averaging 39 weeks. In the 100 mg/kg/day treatment group, 16 boys commenced edasalonexent either at the beginning of Phase 2 or at the beginning of the open-label extension. At the time of the open-label extension data analysis, all 14 boys continuing to participate had received 100 mg/kg/day for 24 weeks and 11 had completed 36 weeks of 100 mg/kg/day edasalonexent treatment. Results are detailed for the 100 mg/kg/day treatment group as all boys are now taking this dose in the open-label extension.
Additional supportive measures of muscle health were also consistent with a positive edasalonexent treatment effect. Four muscle enzymes (creatine kinase, alanine aminotransferase, aspartate aminotransferase and lactate dehydrogenase) were significantly decreased compared to baseline following edasalonexent treatment at 12 weeks and later time points (p<0.05), consistent with the ability to slow muscle degeneration and improve muscle integrity. Rate of change in lower leg MRI T2 significantly improved at 12 weeks and at last observation on treatment compared to control period (p≤0.05), consistent with a reduction of inflammation in the muscle.
Edasalonexent continued to be well tolerated with no safety signals observed to date in the MoveDMD trial. The majority of adverse events (AEs) have been mild in nature with no serious AEs. There have been no dose reductions and no drug-related discontinuations. The most common AEs were gastrointestinal, primarily mild and transient diarrhea. Height, weight and BMI growth patterns were similar to standard growth curves for unaffected boys in the age range of MoveDMD subjects. Boys with DMD in this age range typically have resting tachycardia, a heart rate that exceeds the normal resting rate, and heart rate of the boys treated with edasalonexent decreased toward age-normative values during treatment. We believe these clinical heart rate observations are intriguing and warrant follow-up.
Catabasis plans to commence a single global Phase 3 trial in DMD in the first half of 2018 to evaluate the efficacy and safety of edasalonexent for registration purposes. The planned design of the randomized, double-blind, placebo-controlled trial is informed by discussions with FDA. Catabasis plans for the Phase 3 trial to have many elements in common with the Phase 2 trial including the patient population and endpoints. The trial is anticipated to enroll approximately 125 patients ages 4 to 7 who have not been on steroids for at least 6 months. The primary efficacy endpoint will be change in the North Star Ambulatory Assessment score after 12 months of treatment with edasalonexent compared to placebo. Key secondary endpoints are planned to include age-appropriate timed function tests. Catabasis expects to report top-line results from this trial in 2020.
“We are excited to see positive effects on muscle function with edasalonexent, as we know from research that effects on muscle function are the most important aspect of a therapy for Duchenne for the affected patients and their families,” said Pat Furlong, Founding President and Chief Executive Officer of Parent Project Muscular Dystrophy (PPMD). “We look forward to learning more about edasalonexent as Catabasis begins its Phase 3 trial. With the disease-modifying effects and safety and tolerability profile observed to date for edasalonexent, it has the potential to be a foundational therapy for all people affected by Duchenne.”
Read more.


